NeuroML importer¶
This is a short overview of the nmlimport package, providing
functionality to import a morphology – including some associated information about
biophysical properties and ion channels – from an .nml file.
NeuroML is a XML-based description that provides a common data format for defining and exchanging descriptions of neuronal cell and network models (NeuroML project website).
Working example¶
As a demonstration, we will use the nmlimport library to generate a morphology and
extract other related information from the pyr_4_sym.cell.nml nml file. You
can download it from OpenSourceBrain’s ACnet2 project.
from brian2tools.nmlimport.nml import NMLMorphology
nml_object = NMLMorphology('pyr_4_sym.cell.nml', name_heuristic=True)
This call provides the nml_object that contains all the information
extracted from .nml file. When name_heuristic is set to True,
morphology sections will be determined based on the segment names. In
this case Section names will be created by combining names of the inner
segments of the section. When set to False, all linearly connected
segments combine to form a section with the name sec{unique_integer}.
To obtain a
Morphologyobject:
>>> morphology = nml_object.morphology
|
With this
With this Morphology object, you can use all of Brian’s functions to get information
about the cell:
>>> print(morphology.topology())
-| [root]
`-| .apical0
`---| .apical0.apical2_3_4
`-| .apical0.apical1
`-| .basal0
`-| .basal0.basal1
`-| .basal0.basal2
>>> print(morphology.area)
[ 1228.36272755] um^2
>>> print(morphology.coordinates)
[[ 0. 0. 0.]
[ 0. 17. 0.]] um
>>> print(morphology.length)
[ 17.] um
>>> print(morphology.distance)
[ 8.5] um
You can plot the morphology using brian2tool’s
plot_morphologyfunction:
from brian2tools.plotting.morphology import plot_morphology
plot_morphology(morphology)
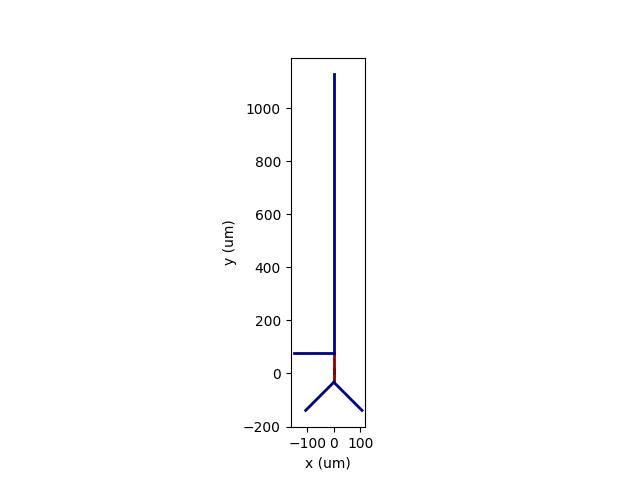
Pyramidal cell’s Morphology plot.¶
Handling SegmentGroup¶
In NeuroML, a SegmentGroup groups multiple segments into a single object, e.g. to
represent a specific dendrite of a cell. This can later be used to apply operations
on all segments of a group. The segment groups are stored in a dictionary, mapping the
name of the group to the indices within the Morphology. Note that these indices
are often identical to the id values used in the NeuroML file, but they do not have
to be.
>>> from pprint import pprint
>>> pprint(nml_object.segment_groups)
{'all': [0, 1, 2, 3, 4, 5, 6, 7, 8],
'apical0': [1],
'apical1': [5],
'apical2': [2],
'apical3': [3],
'apical4': [4],
'apical_dends': [1, 2, 3, 4, 5],
'background_input': [7],
'basal0': [6],
'basal1': [7],
'basal2': [8],
'basal_dends': [8, 6, 7],
'basal_gaba_input': [6],
'dendrite_group': [1, 2, 3, 4, 5, 6, 7, 8],
'middle_apical_dendrite': [3],
'soma': [0],
'soma_group': [0],
'thalamic_input': [5]}
The file pyr_4_sym.cell.nml will look something like this:
1<cell id="pyr_4_sym">
2 <morphology id="morphology_pyr_4_sym">
3 <segment id="0" name="soma">
4 <proximal x="0.0" y="0.0" z="0.0" diameter="23.0"/>
5 <distal x="0.0" y="17.0" z="0.0" diameter="23.0"/>
6 </segment>
7 ..........
8 ..........
9 ..........
10
11 <segment id="6" name="basal0">
12 <parent segment="0" fractionAlong="0.0"/>
13 <proximal x="0.0" y="17.0" z="0.0" diameter="4.0"/>
14 <distal x="0.0" y="-50.0" z="0.0" diameter="4.0"/>
15 </segment>
16 ..........
17 ..........
18 ..........
19
20 <segmentGroup id="apical_dends">
21 <include segmentGroup="apical0"/>
22 <include segmentGroup="apical2"/>
23 <include segmentGroup="apical3"/>
24 <include segmentGroup="apical4"/>
25 <include segmentGroup="apical1"/>
26 </segmentGroup>
27
28 <segmentGroup id="middle_apical_dendrite">
29 <include segmentGroup="apical3"/>
30 </segmentGroup>
31 ........
32 ........
33 ........
34 </morphology>
35</cell>
Handling sections not connected at the distal end¶
If you look at the line 12 in above .nml file, you can see
fractionAlong=0.0. The fractionAlong value defines the point at which the
given segment is connected with its parent segment. So a fractionAlong value
of 1 means the segment is connected to bottom (distal) of its parent segment, 0
means it is connected to the top (proximal) of its parent segment. Similarly a
value of 0.5 would mean the segment is connected to the middle point of its parent
segment. Currently, the nmlimport library supports fractionAlong value to be
0 or 1 only, as there is no predefined way to connect a segment at
some intermediate point of its parent segment in Brian.
Extracting channel properties and Equations¶
The generated nml_object contains several dictionaries with biophysical information
about the cell:
properties:A dictionary with general properties such as the threshold condition or the intracellular resistivity. The names are chosen to be consistent with the argument names in
SpatialNeuron, in many cases you should therefore be able to directly pass this dictionary:neuron = SpatialNeuron(..., **nml_object.properties)channel_properties:A dictionary of reversal potentials and conductance densities for the different channels in the cell. The dictionary maps the name of segment groups to the name of the respective variable names (e.g.
g_Na,E_Na, …)
For the example file, these dictionaries look like this:
>>> pprint(nml_object.properties) # threshold, refractory etc.
{'Cm': 2.84 * ufarad / cmetre2,
'Ri': 0.2 * kohm * cmetre,
'refractory': 'v > 0. * volt',
'threshold': 'v > 0. * volt'}
>>> pprint(nml_object.channel_properties) # reversal potentials and conductance densities
{'all': {'E_LeakConductance_pyr': -66. * mvolt,
'g_LeakConductance_pyr': 0.1420051 * msiemens / cmetre2},
'soma_group': {'E_Ca_pyr': 80. * mvolt,
'E_Kahp_pyr': -75. * mvolt,
'E_Kdr_pyr': -75. * mvolt,
'E_Na_pyr': 55. * mvolt,
'g_Ca_pyr': 10. * msiemens / cmetre2,
'g_Kahp_pyr': 2.5 * msiemens / cmetre2,
'g_Kdr_pyr': 80. * msiemens / cmetre2,
'g_Na_pyr': 120. * msiemens / cmetre2}}
If you have a SpatialNeuron neuron with equations for all the channels in the NML file (e.g. as generated by the
get_channel_equations method below), then you can assign the reversal potentials and conductances to
the individual compartments by combining the information from the segment_groups and
channel_properties dictionaries:
for segment_group, values in nml_object.channel_properties.items():
indices = nml_object.segment_groups[segment_group]
for property, value in values.items():
getattr(neuron, property)[indices] = value
To get the channel equations for a particular channel, e.g. Na_pyr:
>>> print(nml_object.get_channel_equations("Na_pyr"))
alpha_h_Na_pyr = (128. * hertz) * exp((v - (-43. * mvolt))/(-18. * mvolt)) : Hz
beta_h_Na_pyr = (4. * khertz) / (1 + exp(0 - (v - (-20. * mvolt))/(5. * mvolt))) : Hz
I_Na_pyr = g_Na_pyr*m_Na_pyr**2*h_Na_pyr*(E_Na_pyr - v) : A/(m^2)
alpha_m_Na_pyr = (1.28 * khertz) * (v - (-46.9 * mvolt)) / (4. * mvolt) / (1 - exp(- (v - (-46.9 * mvolt)) / (4. * mvolt))) : Hz
beta_m_Na_pyr = (1.4 * khertz) * (v - (-19.9 * mvolt)) / (-5. * mvolt) / (1 - exp(- (v - (-19.9 * mvolt)) / (-5. * mvolt))) : Hz
dh_Na_pyr/dt = alpha_h_Na_pyr*(1-h_Na_pyr) - beta_h_Na_pyr*h_Na_pyr : 1
dm_Na_pyr/dt = alpha_m_Na_pyr*(1-m_Na_pyr) - beta_m_Na_pyr*m_Na_pyr : 1
E_Na_pyr : V (constant)
g_Na_pyr : S/(m^2) (constant)
Note that this mechanism currently only supports passive channels and HH-type channels with sigmoidal, exponential, or exponential linear equations for the gating variables.
Note
If your .nml file includes other .nml files, make sure they
are present in the same folder as your main .nml file. If the files are
not present, a warning will be thrown and execution will proceed as normal.